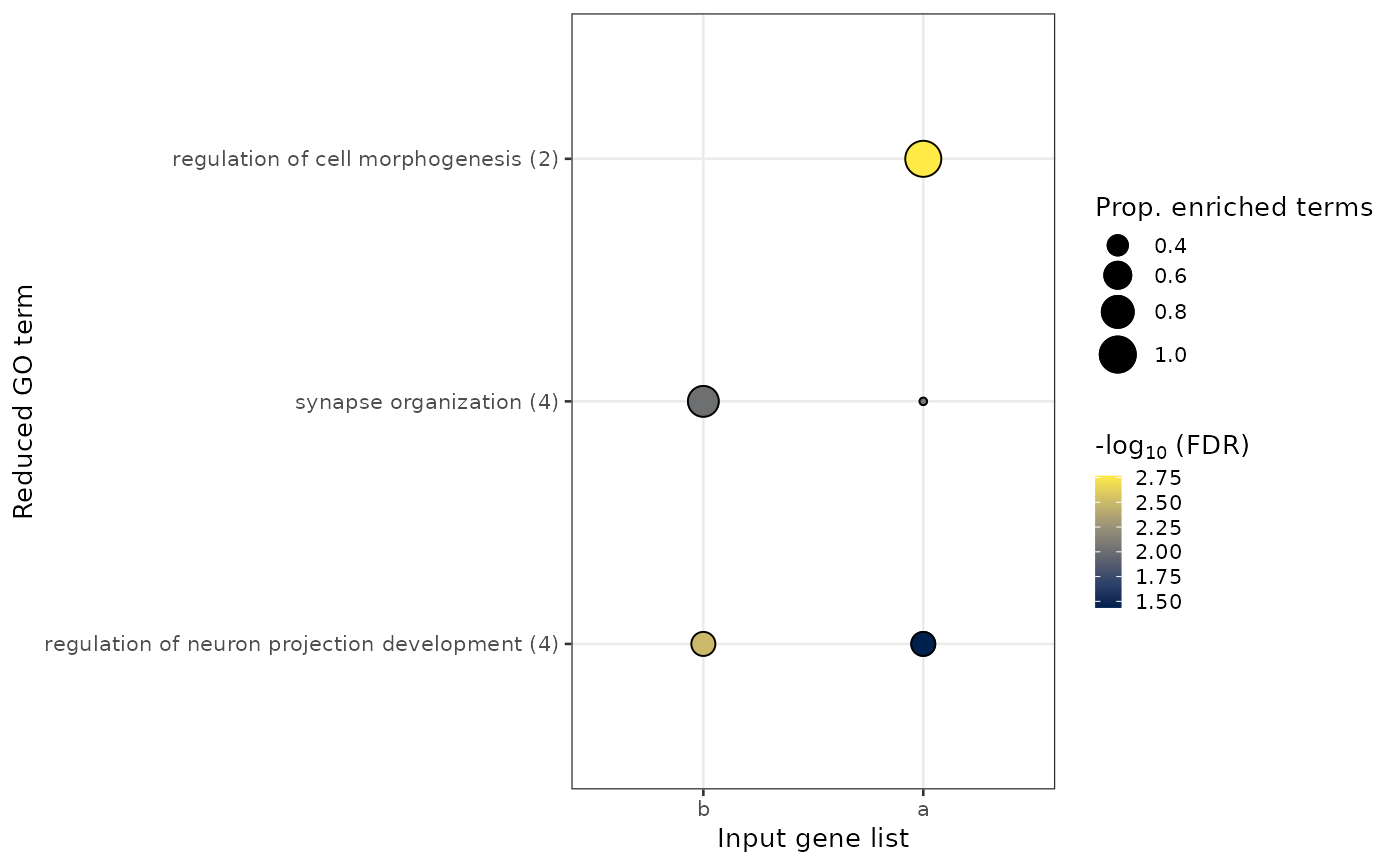
go_plot plots GO terms that have been reduced using go_reduce.
go_plot( reduced_pathway_df, col_gene_list = "gene_list", col_fdr = "fdr", fct_gene_list = NULL )
Arguments
| reduced_pathway_df | |
|---|---|
| col_gene_list | a |
| col_fdr | a |
| fct_gene_list | a |
Value
ggplot displaying the pathway enrichments using reduced perent
terms.
x-axis displays the inputted gene list(s).
y-axis displays the reduced parent term, with the number of associated child GO terms in brackets. Parent terms are ordered first by the factoring of the input gene list (as provided in the argument,
fct_gene_list) and then by the number of associated child terms.size of the dot indicates the proportion of child terms associated with an inpute gene list that are annotated to the parent term.
fill of dot indicates the minimum FDR assigned to a child term that is associated with the parent term (within an input gene list).
See also
Examples
file_path <- system.file( "testdata", "go_test_data.txt", package = "rutils", mustWork = TRUE ) pathway_df <- readr::read_delim(file_path, delim = "\t" ) %>% dplyr::mutate( gene_list = c(rep("a", 5), rep("b", 5)), fdr = runif(10, min = 0, max = 0.05) )#> #>reduced_pathway_df <- go_reduce( pathway_df = pathway_df, threshold = 0.9, scores = NULL, measure = "Wang" )#> [1] "Reducing sub-ontology: BP"#>#>#>#>#>#>#> #>#> #> #> #> #>#> #> #>#> #> #> #> #> #> #> #>#>#> #> #> #> #>#>#>#> #>#> #> #>#>#>#>go_plot( reduced_pathway_df, col_gene_list = "gene_list", col_fdr = "fdr", fct_gene_list = c("b", "a") )#>